PolyA-miner
More than half of human genes undergo alternative polyadenylation (APA) and generate mRNA
transcripts with varying lengths. Increasing awareness of APA’s role in human health and disease
has propelled the development of several 3’ sequencing (3’Seq) techniques. Despite the recent
data explosion, computational tools that are precisely designed for 3’Seq data are not well
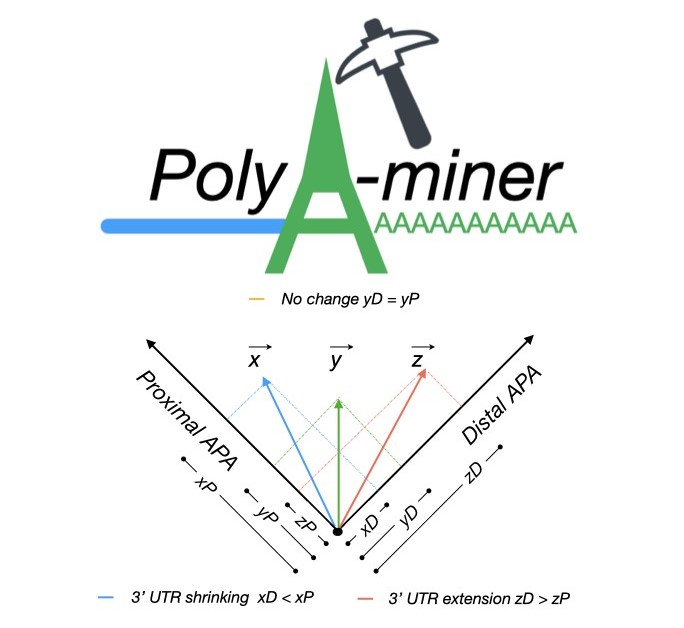
established. PolyA-miner is developed specifically for 3’Seq data, it accounts for all
non‐proximal to non‐distal APA switches using vector projections and reflect precise gene level
3’UTR changes. PolyA-miner is less susceptible to inherent data variations can also to
effectively identify novel APA sites that are otherwise undetected using reference-based
approaches. With the emerging importance of alternative polyadenylation in studying human
diseases, PolyA-miner can significantly accelerate data analysis and help decoding the missing
pieces of underlying alternative polyadenylation dynamics.
Read
More
CrypSplice
Alternative splicing of RNA is the key mechanism by which a single gene codes for multiple
functionally diverse proteins. Several studies established compromised RNA homeostasis (splicing
errors) and identified previously unknown class of exons, ‘cryptic’ exons, in RNA transcripts.
These cryptic exons are often associated with various neurological disorders and cancers.
Genome-wise detection of cryptic splice sites can facilitate a comprehensive understanding of
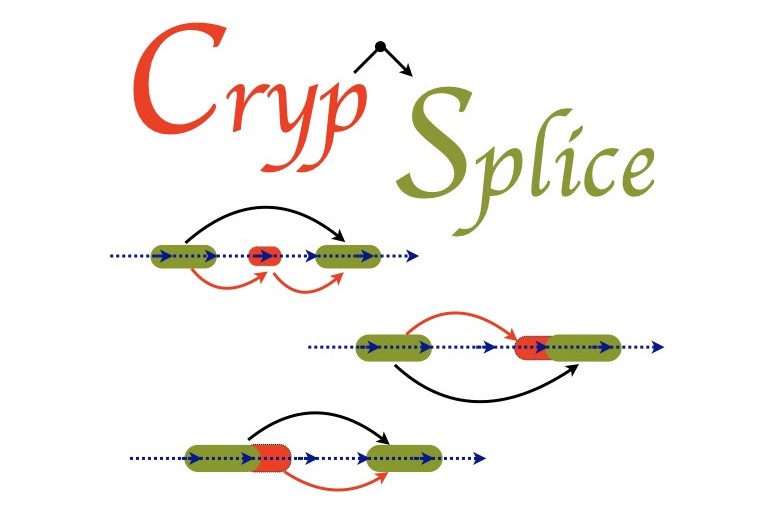
the underlying disease mechanisms and develop therapeutic strategies. CrypSplice can effectively
quantify and evaluate cryptic splicing patterns from RNASeq data using a beta‐binomial
distribution model. CrypSplice, revealed extensive cryptic slicing in Amyotrophic lateral
sclerosis and Spinocerebellar ataxia models.
Read More
SpliceNet
Conventionally total gene expressions are used to infer gene networks, but it is challenging to
account splicing isoforms, especially disease specific isoforms. RNA sequencing made splice
variant profiling practical. However, its true merit in quantifying splicing isoforms and
isoform‐specific exon expressions is not well realized in inferring gene networks. Addressing
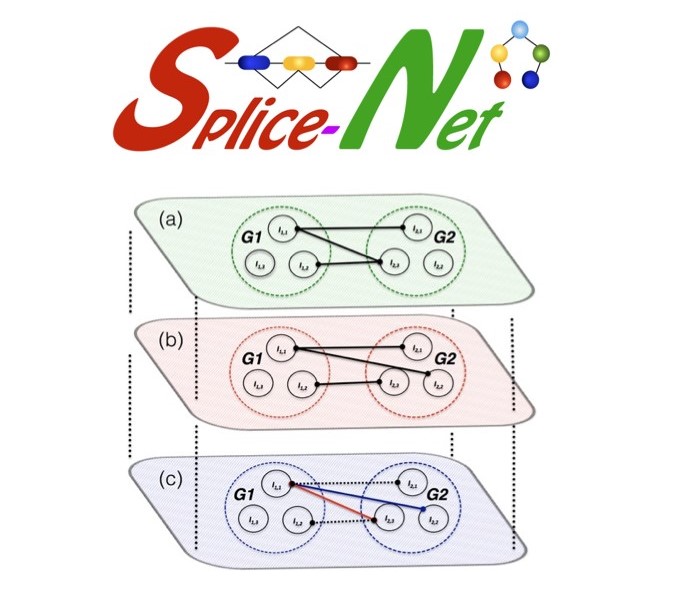
this SpliceNet infer isoform‐specific co‐expression networks from exon‐level RNA‐Seq data. It
goes beyond genes and infer disease specific splicing isoform networks. SpliceNet revealed
several novel isoform co‐expression patterns in TCGA lung cancer data.
Read More
DDGni
Inferring gene‐regulatory networks is very essential to understand any biological systems.
However, the complexity of transcription and translation induces delay in gene regulation and
this delay is often dynamic, making network inference hard. Although a number of
gene‐network‐inference methods are proposed, most of them ignore the associated dynamic time
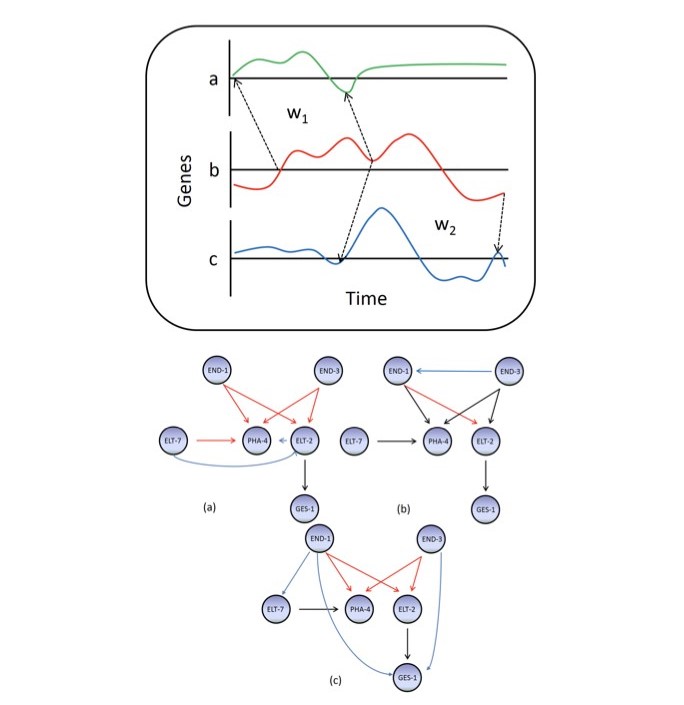
delay. I targeted this specific problem and developed DDGni a dynamic delay gene‐network
inference algorithm based on local dynamic time wrapping technique that can auto tune to the
delay dynamics in gene regulation. I have successfully demonstrated the merit of DDGni on both
synthetic and real time live cell expression data.
Read More